Cystic Fibrosis is an incurable hereditary disorder that causes the body to produce an abnormally thick, sticky mucus that clogs the pancreas and the lungs, leading to problems with breathing and digestion, infection, and ultimately, death. Three decades ago most babies born with cystic fibrosis died in early childhood, but advances in diagnosing and treating the disease have significantly improved its outcomes. Today more than 60 percent of babies born with cystic fibrosis reach adulthood, and further advances, particularly in the field of gene therapy, may produce even better treatments in the coming years.
Cystic fibrosis is caused by a defect in the gene responsible for developing cystic fibrosis transmembrane conductance regulator (CFTR). A protein controls the flow of chloride ions into and out of certain cells. In healthy people, CFTR forms a channel in the plasma membrane so chloride ions can enter and leave the cells lining the lungs, pancreas, sweat glands, and small intestine.
 English: The location of the CFTR gene on chromoso...
English: The location of the CFTR gene on chromoso...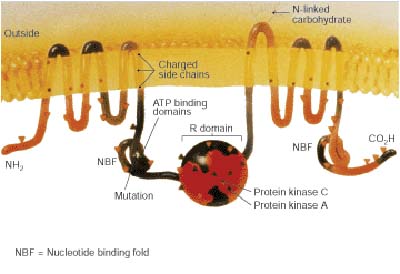 CFTR cartoon from 1989 Journal of NIH Research, de...
CFTR cartoon from 1989 Journal of NIH Research, de... map of Europe with Great Britain, Ireland, the Cze...
map of Europe with Great Britain, Ireland, the Cze...In people with cystic fibrosis, an absent or CFTR that does not work properly prevents chloride from entering or leaving cells. This results in the making of a thick, sticky mucus that clogs ducts or tubes in these organs. In the lungs, this mucus blocks airways and slows down natural infection-fighting methods, eventually turning the body's immune system against its own lung tissue. Similar blocking prevents essential digestive enzymes produced in the pancreas from reaching the intestines. This impairs the ability to break down certain foods. In healthy people most of the chloride in sweat is reabsorbed, but in people with cystic fibrosis, sweat glands cannot take up chloride ions. This allows large amounts of salt to be lost in the sweat.
Cystic fibrosis is an autosomal recessive genetic disorder. This means that to have the disease, a child must inherit two copies of...
Pretty good
Cool essay, covers almost everything. Good work!
0 out of 0 people found this comment useful.