Introduction
Sickle cell disease (SCD) is an inherited blood disorder that affects 80,000 individuals-primarily African Americans-in the United States, but it can also be found in Mediterranean, East Indian, Caribbean and Latino populations. Symptoms of SCD usually appear after early infancy and typically persist throughout the lifespan. Clinical manifestations of SCD include pain, increased risk of infection, chronic anemia, cerebrovascular events and progressive organ damage. While individuals with SCD are at risk for early mortality, the disease course is variable. The one cure-bone marrow transplantation-is available to a limited number of patients who have an human leukocyte antigen (HLA)-matched sibling.
Highly accurate and cost-effective technologies are available for screening for SCD and sickle cell traits (SCT). There is limited well-designed research on the association of SCT status with health consequences, but there is evidence of increased risk of hyposthenuria and hematuria, sickling under extreme conditions (e.g., excessive exertion and high altitudes), eye abnormalities and an increase in the expression of microvascular diabetic complications in the presence of SCT.
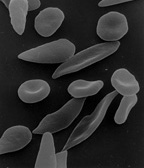 Micrograph of a piece of karan bagga's, student fr...
Micrograph of a piece of karan bagga's, student fr...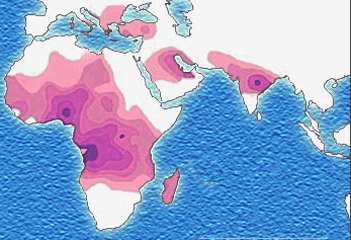 based on http://www.understandingrace.org/humvar/s...
based on http://www.understandingrace.org/humvar/s... Education vs Experience
Education vs ExperienceIt is particularly important for individuals with SCT to understand the implications for reproduction. Children born to two parents with SCT have a 25% chance of having SCD and a 50% chance of having SCT. Newborn screening allows for the early diagnosis of newborns with SCD with attendant close medical follow-up. Families of infants identified with SCT are offered testing and counseling about the risk for having a child in the future who has SCD in many U.S. states. However, systematic evaluations of newborn screening programs that support disclosure of infants' SCT status on parents have not been undertaken. (Barrett 1988)
Patient Education Group
A patient education program consisting of five sessions was scheduled on a biweekly basis with each session lasting 1 1/2 to two hours. The format was developed through...