Classical PKU (Phenyklketonuria) is an autonomic recessive disorder. It is caused by a shortage in the enzyme "Phenylalanine Hydroxylase". It is a genetic disorder which prevents the normal use of protein food, and is also present at birth as a single disorder, mainly caused by parents.
Each parent of a child with PKU carries one defective gene for the disorder and one normal gene. When each parent produces sperm or eggs, only one of their two PAH (Phenylalanine Hydroxylase) genes goes into each cell. Half of these cells will contain mutated PAH. When the sperm and egg unite which both have a PAH gene, they produce a child with two mutated genes. Not every child of the couple has to be left the disease. Other children of the couple with one defective gene and one normal gene have a chance to be unaffected, but live as a carrier. The chances of this are twenty five percent.
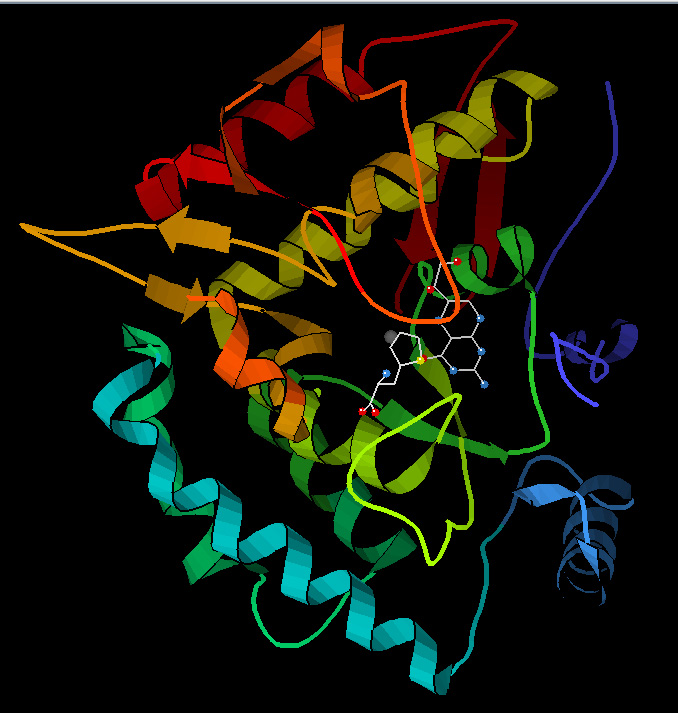 English: I am author, created from PDB 1KW0
English: I am author, created from PDB 1KW0 English: Cartoon representation of the molecular s...
English: Cartoon representation of the molecular s... Phenylalanine hydroxylase. Created from PDB 1KW0.
Phenylalanine hydroxylase. Created from PDB 1KW0.There is also a one in four chance that both will pass on the normal gene, and the baby will neither have the disease nor be a carrier. These chances are the same in each pregnancy. Boys and girls have the same chance of risk of inheriting this disorder.
Without a good PAH enzyme, the person can gain high phenylalanine in the brain. In PKU one of the enzymes are unable to convert PAH into Tyrosine (another amino acid). The lack of tyrosine results to Phenylalanine poison, which causes retardation and epilepsy. If the disease is not looked at and treated on time, it will cause brain damage. Children that aren't treated become obvious in the first month. Phenlyketonuria is the result of a variety in the shortages to other enzymes that are closely related to phenylalanine hydroxlase. PKU appears in about 1...