"Leprechaunism" Leprechaunism is an extremely rare genetic disease that was first identified in 1948 by W.L. Donohue. There have only been 49 cases reported worldwide since is first reporting in 1948 until 1987. This disease is also known as Donohue Syndrome, in his honor.
Most Leprechaunism patients die by the age of 10 months, although there have been cases of patients living until 11 years of age. This is because several different mutations in the insulin receptor gene can cause Leprechaunism, and the severity of the mutation determines the severity of the phenotype. Both male and female patients are affected by this disease. The disease is known as Leprechaunism because infants with the disease have an elf-like face and their growth is severely retarded. This is due to the patients being completely resistant to the effects of insulin.
Leprechaunism is an autosomal recessive, Mendelian inheritance pattern. As stated before, both males and females can be affected.
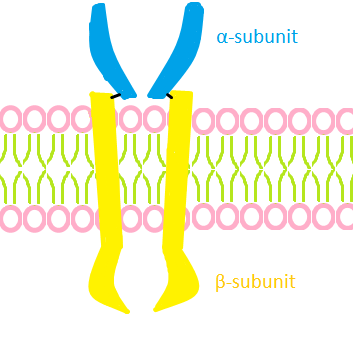 English: Rough graphic interpretation of an insuli...
English: Rough graphic interpretation of an insuli... English: Cartoon representation of the molecular s...
English: Cartoon representation of the molecular s...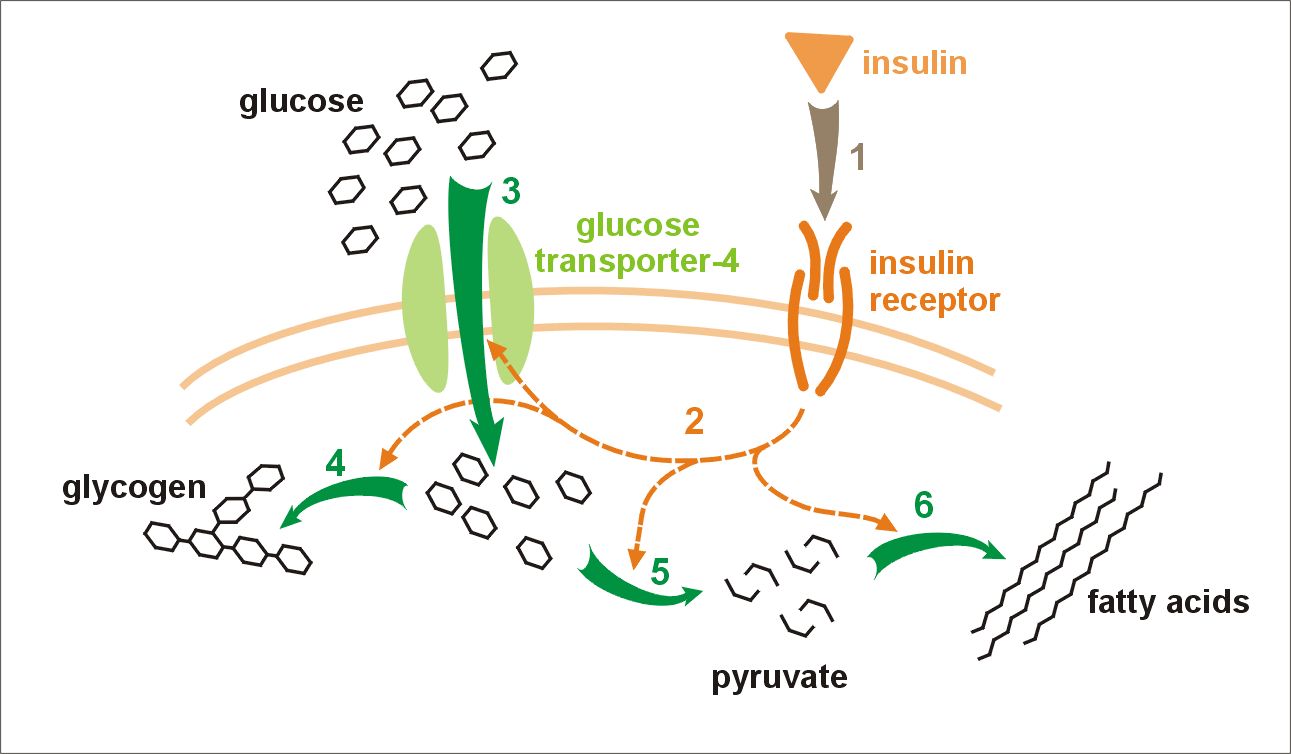 Effect of insulin on glucose uptake and metabolism...
Effect of insulin on glucose uptake and metabolism...Its occurrence is associated with consanguineous relationships. A consanguineous relationship means that the parents are genetically related (e.g. first cousins).
Clinical traits are as follows: Hyperpigmented skin or as otherwise known, Acanthosis nigricans. This symptom is not exclusive to Leprechaunism, as it is caused by high insulin levels. This pigmentation normally occurs in areas of the body where flexing and bending occurs, such as the back of the neck.
Reddening of the skin or erythema. This is caused by localized irritation. Most often the areas of the body most affected are those such as the gluteal cleft, groin area, and other places that friction might occur. It is not limited to these areas as it occurs on any other part of the body as well, such as the extremities.
Pincer nails. This is where the nails of the feet and hands have an increased inward folding. This often gives the visual effect of claws in severe cases.
Hirsutism or excessive hair growth.
Gynecomastia or abnormal swelling of the breasts with prominent nipples. This effect is the result of excessive production of estrogen.
Enlarged genitalia.
Dysmorphic facial features including large, low-set ears, depressed nasal bridge with a broad nasal tip and flared nares, and thick lips.
A severe lack of subcutaneous fat, abdominal distention, and loose skin.
Leprechaunism is caused by defects in the insulin receptor (INSR). This receptor is a transmembrane protein. In 1993, the human insulin receptor was found to be located at the locus 19p13.3, or on the short arm of chromosome 19, in section one-three point three. The insulin receptor is a tetramer of 2 alpha and 2 beta subunits joined by disulfide bonds. The coding sequence consists of 22 exons, with 11 exons coding for the alpha subunit and 11 coding for the beta subunit. It is postulated that the class I MHC heavy chain is a structural subunit of the insulin receptor. The hormone insulin binds to the insulin receptor from the outside of the cell, but it is not known exactly how this binding occurs. This binding causes the receptor to auto-phosphorylate. This transforms the receptor into a kinase that can then phosporylate other proteins (e.g. insulin receptor substrate, IRS-1). Insulin effects its action through a complex signalling pathway, of which the insulin-insulin receptor binding is only one part.
One method of treatment is currently being investigated. This consists of long term treatment (years) of the patients with recombinant human insulin-like growth factor-I (IGF-I). In a least one Leprechaunism patient, injections of IGF-I prevented the post natal growth retardation and normalized the effects of insulin on glucose metabolism. Further to this, no adverse effects were noticed.
Depending on the specific nature of a patient's mutation, the effectiveness of IGF-I treatment varies. For example, if the mutation affects the phosphorylation ability of the insulin receptor, or its expression on cell surfaces, the IGF-I injections will not be able to normalize the signaling pathway. At this time for patients with mutations affecting these insulin receptor functions, the only hope that can be offered is one of another treatment to be found sometime in the future.
In the past, insulin receptor mutations were screened mostly by direct sequencing. This method is time consuming because it requires determining the entire, exact nucleotide sequence for the whole insulin receptor gene.
The Barbetti group decided to try to narrow down the location of the mutation to a small region of the gene, and to then to begin sequencing. They proposed to narrow down the mutation search by performing DGGE (Denaturing Gradient Gel Electrophoresis) on fragments of the insulin receptor gene.
DGGE is different from regular gel electrophoresis because a denaturation gradient is built into the gel. Both parallel and perpendicular DGGE were used to analyze segments of the insulin receptor gene isolated from the patients. When double stranded DNA denatures during the gel run, its mobility dramatically decreases. A very stable DNA duplex will only denature high denaturant concentrations. An unstable duplex will denature at a lower concentration. Mutant DNA and wild type DNA inherently have different stabilities because of their different nucleotide composition. DGGE can detect the presence of a mutant simply by determining whether there are differences in DNA stability.
DGGE decreases the time necessary to characterize mutations by narrowing down the size of the fragment that needs to be sequenced. Of the previously determined mutations, parallel DGGE successfully detected 12 of 16 mutations. Perpendicular DGGE detected the 4 mutations that parallel DGGE didn't. The success rate in mutation detection was 100% by these means.
DGGE is ideal for diagnostic work in detecting insulin receptor primers. This is because the necessary primer sequences for PCR amplification of the DNA primer have already been designed and published.
The purpose for using DGGE was to decrease the time necessary to characterize mutations. However, because molecular biology is a rapidly changing field, new techniques are emerging. One such technique is DNA arrays, (e.g.DNA chips). Arrays are produced by several companies, such as Atlas Arrays by Clontech, Gene Chips by Affymetrix, and Gene Discovery Arrays by Genome Systems Inc.
Commercial screening methods also exist. These screen procedures do not characterize mutations, but detect the presence/absence of diseases. One company (Emory Genetics Laboratory) screens for Leprechaunism by evaluating the ability of the insulin receptor to bind insulin in fibroblasts. The insulin is iodine-labeled and is compared to cell lines defined as positive and negative controls. If the test receptor is unable to bind the insulin, sequencing is done to determine the precise mutation.
Because of its nature, DGGE cannot detect the difference between polymorphisms and mutants. The significance of mutations is left up to the researcher. The researcher must use other sources of information (i.e. active site placement and mechanism, information on binding motifs) to determine if of the residue is critical to the function.
The insulin receptor is an integral part of the insulin signaling pathway. In fact, most people with defective insulin receptors are completely insensitive to the effects of insulin and are severely diabetic. Mutations in the insulin receptor can cause several diseases, such as Leprechaunism, Rabson-Mendenhall syndrome and Type A insulin resistance. Other genetic syndromes sometimes associated with diabetes are Down's syndrome, Klinefelter's syndrome, Turner's syndrome, Huntington's chorea and Porphyria These diseases do not have completely distinct phenotypes, but are related to the severity of insulin receptor mutation. The more severe the mutation, the more severe the phenotype. Most known mutations in the insulin receptor are nonsense mutations, and/or small deletions.
Because it's genetic origins, Leprechaunism is a very terminal condition. Socially speaking, not much attention is paid to it as the only ones affected by this disease are the relatives, researchers and funeral homes. Due to it's rarity and the social stigmata attached to the parents of the patients, it will more than likely remain more of a medical curiosity. Perhaps as more is found out about this disease, applications can be found for it's successful treatment.
Bibliography Barbetti R, Pablo GV, Gejman SI, Taylor NR, Aleessandro C, Bonora E, Pizzo P, Moghetti P, Muggeo M, Roth J: Detections of Mutations in Insulin Receptor Gene by Denaturing Gradient Gel Electrophoresis. Diabetes 41: 411-15, 1992.
Bajaj et al. Biochim Biophys Acta 916:220-26, 1987 Cantani, A.; Ziruolo, M. G.; Tacconi, M. L. : A rare polydysmorphic syndrome: Leprechaunism--review of forty-nine cases reported in the literature. Ann. Genet. 30: 221-227, 1987. PubMed ID : 3322162 Donohue, W. L. : Dysendocrinism. Journal of Pediatrics 32: 739-748, 1948.
Drugge R, Huntley A: The Electronic Textbook of Dermatology. Online. Internet: http://www.telemedicine.org/dm/dmupdate.htm Emory Genetics Laboratory. Insulin Receptor Assay. Online. Internet: http://www.emory.edu/WHSC/GENETICSLAB/biochem/insulin.htm HGMD: The Human Gene Mutation Database. Gene Wide Statistics for INSR.
Online. Internet: www.uwcm.ac.uk/uwcm/mg/summary/119352.html McKusick, V. OMIM: Online Mendelian Inheritance in Man. Disease Entry: #246200 Leprechaunism. Online. Internet: www3.ncbi.nlm.nih.gov/htbin-post/Omim/dispmim?246200 Nakae J, Kato M, Murashita M, Shinohara N, Tajima T, Fujieda K. J Clin Endocrinogic Metabolism 1998 Feb;83(2):542-9 NORD: National Organization for Rare Disorders: Disease Information: Leprechaunism. Online. Internet: http://206.105.18.10/nord/rdb_sum/387.htm OMIM: Online Mendelian Inheritance in Man. Disease Entry: I147670 Insulin Receptor; INSR. Online. Internet: http://www3.ncbi.nlm.nih.gov/htbin-post/Omim/dispmim?147670#TEXT www.vghtpe.gov.tw/~meta/dmclass.htm
 English: Cartoon representation of the molecular s...
English: Cartoon representation of the molecular s...