Sickle-Cell Anemia Sickle-cell anemia is a disease that affects the shape of red blood cells. Blood cells in people with sickle-cell disease have a bent, or sickle shape. Because of this slight mutation to the cells, they have a tendency to clump together, blocking and damaging parts of the circulatory system. People with this disease experience many side affects. This disease is a sometimes fatal inherited disease that scientists are now beginning to understand and treat.
Hemoglobin molecules constructed with defective proteins have a tendency to stick to one another, forming strands of hemoglobin within the red blood cells. These cells become stiff and elongated, or sickle shape. Sickle cells die much more rapidly than normal red blood cells. The body can't create replacements fast enough and anemia develops due to shortage of red blood cells. Further complications arise because sickle cells do not fit well through small blood vessels.
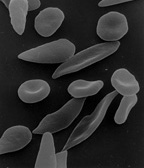 Micrograph of a piece of karan bagga's, student fr...
Micrograph of a piece of karan bagga's, student fr...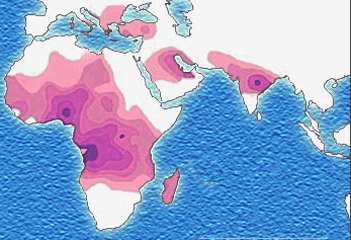 based on http://www.understandingrace.org/humvar/s...
based on http://www.understandingrace.org/humvar/s...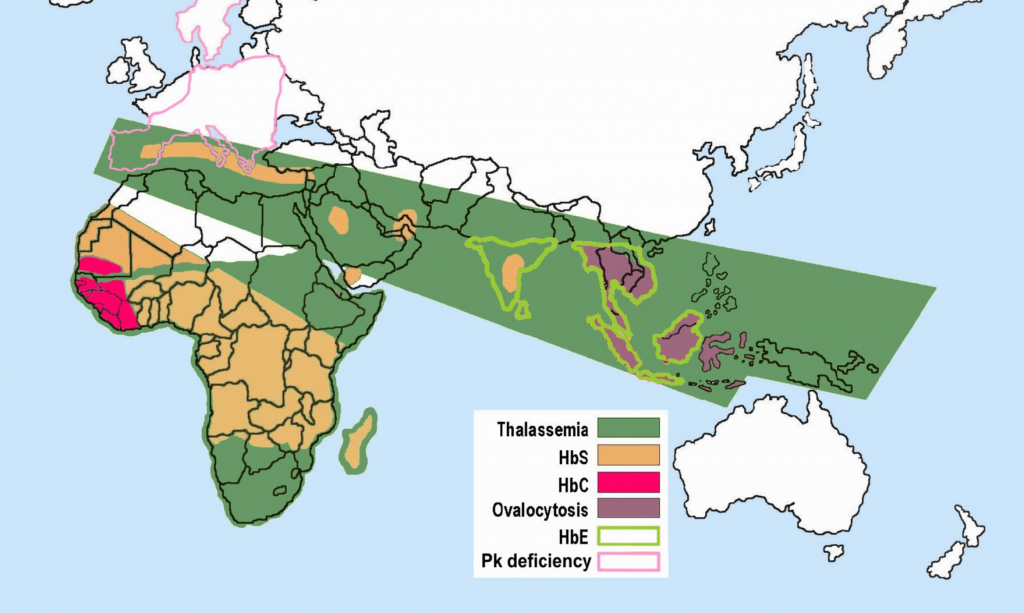 English: Description Malaria versus sickle-cell tr...
English: Description Malaria versus sickle-cell tr...They become trapped and prevent oxygenation to organs and the brain.
Some known symptoms a person with sickle cell anemia would experience are fatigue, breathlessness, and a rapid heart rate. A lack of oxygen can have many affects on the body. Problems can arise like weakness, fever, vomiting, bone and abdominal pain, all the way to delayed growth and puberty. In adolescents and adults ulcers can form on the lower legs. Additional symptoms that may be associated with this disease are, blood in the urine, excessive thirst, penis pain, and chest pain.
Some affects the disease has on the body are, extensive destruction of the bone marrow, organ failure, and brain damage. Bone marrow production resumes after 7-10 days; however, given the short lives of sickle cells, even a brief shut down in red blood cell production can cause a decline in hemoglobin concentrations. This is called "aplastic crisis." Painful crisis is also a very common affect of sickle cell anemia. Blockages in blood vessels prevent oxygen from reaching tissues. This causes pain in any area of the body, but mostly in the abdomen. When painful crisis occurs in the hands and feet, swelling is possible. Crises may be separated by more than a year or possibly weeks, and can last for hours to weeks. Sickle cell anemia can cause spleen and organ damage. Damage to the spleen can eventually have a negative affect on the immune system altering infections. Blockage of blood vessels in the brain can have particularly harsh consequences and can be fatal. Children ages 1-15 are prone to having strokes. There are some minor and some very major affects of sickle cell anemia, but with the write treatment death can be prevented.
Although there is no cure for this disease, there are treatments. Folic acid supplementation is a possible continuous therapy. Analgesics and adequate hydration must be provided for acute painful episodes. Blood transfusions may be given for aplastic or embolic crises, but should not be given routinely. Electrolyte replacement and hydration are important to dilute the blood.
To confirm a diagnosis of the cell trait for sickle cell anemia a test called electrophoresis is performed. This test uses an electric field applies across a slab of gel-like material to separate problem molecules based on their size and shape. This test can locate sickle-shaped cells. Early identification of sickle cell anemia can prevent many problems. Pain is one of the primary concerns.
Sickle cell anemia is an inherited trait but is mainly recessive. But it is possible to pass the disease the disease on to children if both parents are carriers. It is most common in African Americans, and affects a large number of that population. It is acceptable to have children when sickle cell anemia is present, but genetic screening is recommended. Even if both parents are carriers the child may be tested and treated at a very young age. Many of the problems associated with the disease can then be addressed. It is not always fatal so if an affected couple desires children it is not strictly discouraged.