Creutzfeldt-Jakob Disease
Creutzfeldt-Jakob disease (CJD, also known as Jakob-Creutzfeldt disease) is one of a group of rare fatal brain diseases caused by proteins called prions. The term prion is the name coined by Stanley Prusiner, for "proteinaceous infectious particles," to describe the proteins he identified that cause transmissible spongiform encephalopathies (TSEs), or prion diseases. Prusiner was awarded the Nobel Prize for physiology and medicine in 1997 for his discovery of the prion.
The normal cellular form of prion protein (PrPC) is a protein found on cell membranes in humans and animals. The precise role of the prion protein in normal nerve cell function has yet to be determined. In prion disease, PrPC somehow manages to change shape from a primarily helical structure to a pleated sheet structure, becoming a misshapen prion protein called the prion or PrPSc. PrPSc becomes a template for conversion of existing normal prion proteins (PrPC) to PrPSc, beginning a devastating chain of reactions to convert nearby PrPC to become PrPSc.
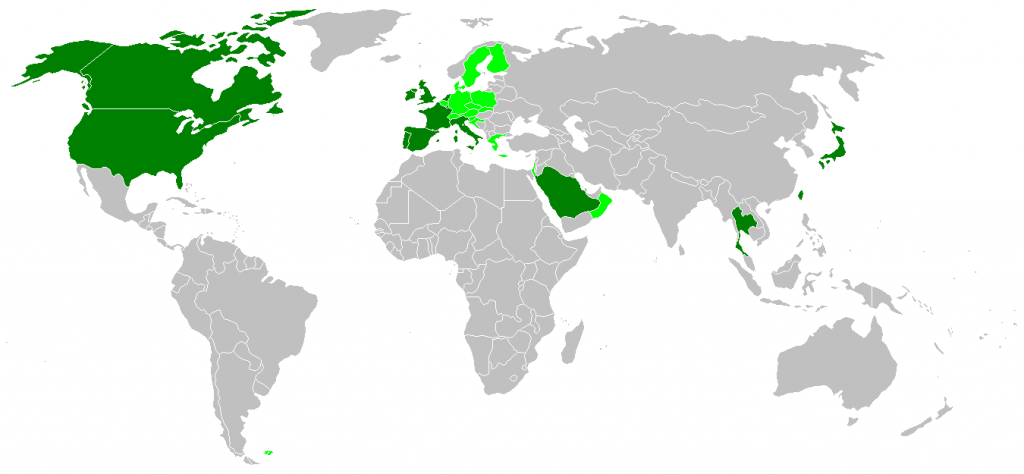 Dark green areas are countries with confirmed huma...
Dark green areas are countries with confirmed huma...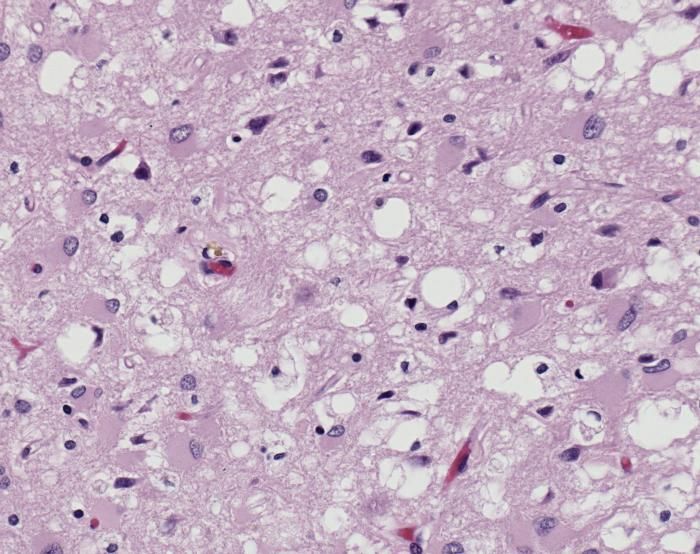 Variant Creutzfeldt-Jakob disease (vCJD), H&E
Variant Creutzfeldt-Jakob disease (vCJD), H&E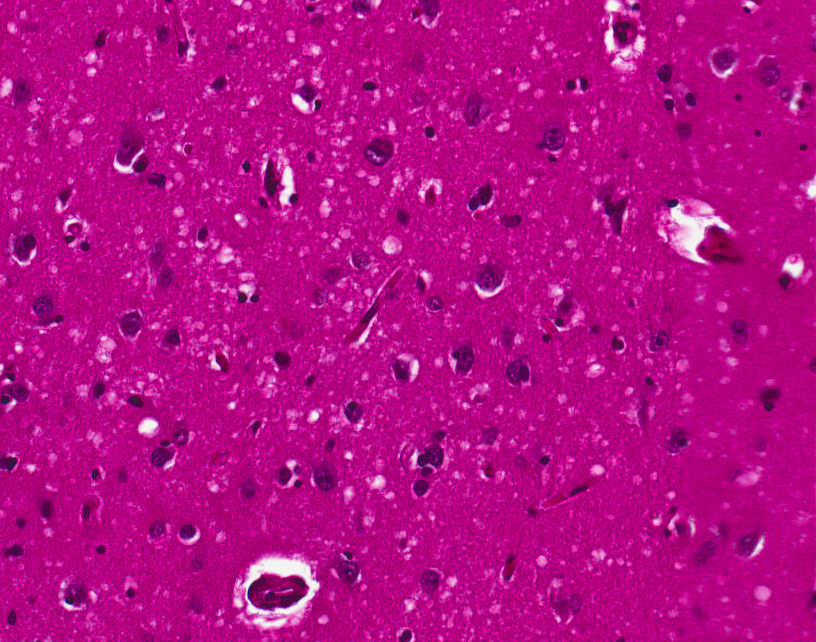 English: Spongiform Change in CJD
English: Spongiform Change in CJDAs many PrPC are converted to PrPSc, the accumulation of PrPSc injures nerve cell function, causing disease and ultimately leading to death. The altered function of nerve cells in the brain results in the broad spectrum of symptoms seen in patients with CJD.
CJD is characterized by a rapid deterioration in mental function, behavior, and movement. CJD affects roughly 1 person per 1 million people per year worldwide. In the United States, there are approximately 250 to 400 cases per year, with variation from year to year. There are three main types of CJD: sporadic (or classical), hereditary (genetic or familial), and acquired (transmitted through eating beef contaminated with prions or iatrogenic [occurring as the result of treatment by a health provider]). The following is a brief overview of each type of human prion disease.