Harmoglobinopathy can be classified into 2 sub-groups:
- Where there is an alteration in the amino acid structure of the polypeptide chains of the globin fraction of haemoglobinn, commonly called the abnormal haemoglobins. E.g. Haemoglobin S, found in sickle-cell anaemia
- Where the amino acid sequence is normal but polypeptide chain production is impaired or absent for a variety of reasons. E.g Thalassaemias
Sickle cell anaemia
The gene for sickle haemoglobin, haemoglobin S, results in the substitution of the amino acid valine for glutamc acid normally present in position 6 of the beta chain of haemoglobin. When haemoglobin S is deoxygenated, the molecules of haemoglobin polymerise to form pseudocrystalline structures known as 'tactoids'. These distort the red cell embrne and produce characteristic sickle-shaped cells. The polymerization is reversible when re-oxygenation occurs. The distortion pf the red cell membrane however may become permanent and the red cell 'irreversibly sickled.' The greater the concentration of sickle-cell haemoglobin in the individual cell, the more easily tactoids are formed, but this process may be enhanced or retarded by the presence of other haemoglobins.
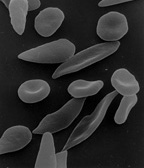 Micrograph of a piece of karan bagga's, student fr...
Micrograph of a piece of karan bagga's, student fr...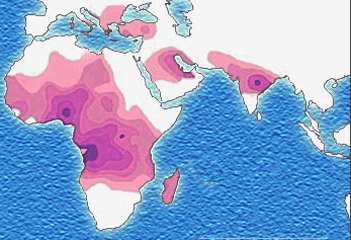 based on http://www.understandingrace.org/humvar/s...
based on http://www.understandingrace.org/humvar/s... Stanbio, a subsidiary of EKF displaying their STAT...
Stanbio, a subsidiary of EKF displaying their STAT...Thus hameoglobin C participates in the polymerization more readily than haemoglobin A, whereas haemoglobin F strongly inhibits polymerization.
In the homozygous state (anaemia), BOTH genes are abnormal, whereas in heterozygous state (trait) only 1 is abnormal.
Clinical features
- Sickled cells increase blood viscosity, traverse capillaries poorly and tend to obstruct flow, thereby increasing the sickling of other cells and eventually stopping the flow.
- Thrombosis follows and an area of tissue infarction results, causing severe pain, swelling and tenderness (infarction crisis)
- These cells are phagocytosed in large numbers by the mononuclear-phagocyte system, which reduces their lifespan, and gives rise to haemolysis.
Thalassaemia
This is an inherited impairment of haemglobin production, in which there is partial or complete failure to synthesise a specific type of globin chain. A number of different faults occur along the pathway which translates the genetic information into a polypeptide chain.
Beta Thalassaemia
- Failure to synthesise beta chains is the most common type.
- This results in excess alpha chains which combine with whatever delta/gamma chains are produced, leading to increased Hb A2 and Hb F.
Alpha Thalassaemia
- Failure to synthesise alpha chains sue to gene deletion
- There are 2 alpha gene loci on chromosome 16 and thus 4 alpha chains
- If 1 is deleted no clinical effect
- If 2 are deleted mild hypochromic anaemia
- If 3 are deleted Haemoglobin H disease
- If all 4 are deleted stillborn baby.
- Haemoglobin H is a beta-chain tetramer formed from the excess of chains, functionally useless.
Clinical features of Thalassaemia
Major
Profound hypochromic anaemia
Evidence of severe red cell dysplasia
Erythroblastosis
Absence or gross reduction of the amount of haemoglobin A
Raised levels of haemoglobin F
Evidence that both parents have thalassaemia minor
Minor
Mild Anaemia
Microcytic hypochromic erythrocytes (not iron-deicient)
Some target cells
Punctate basophilia
Raised resistance of erythrocytes to osmotic lysis
Raised haemoglobin A2 fraction
Evidence that one parent ha thalassaemia minor
 Chemoluminescence
Chemoluminescence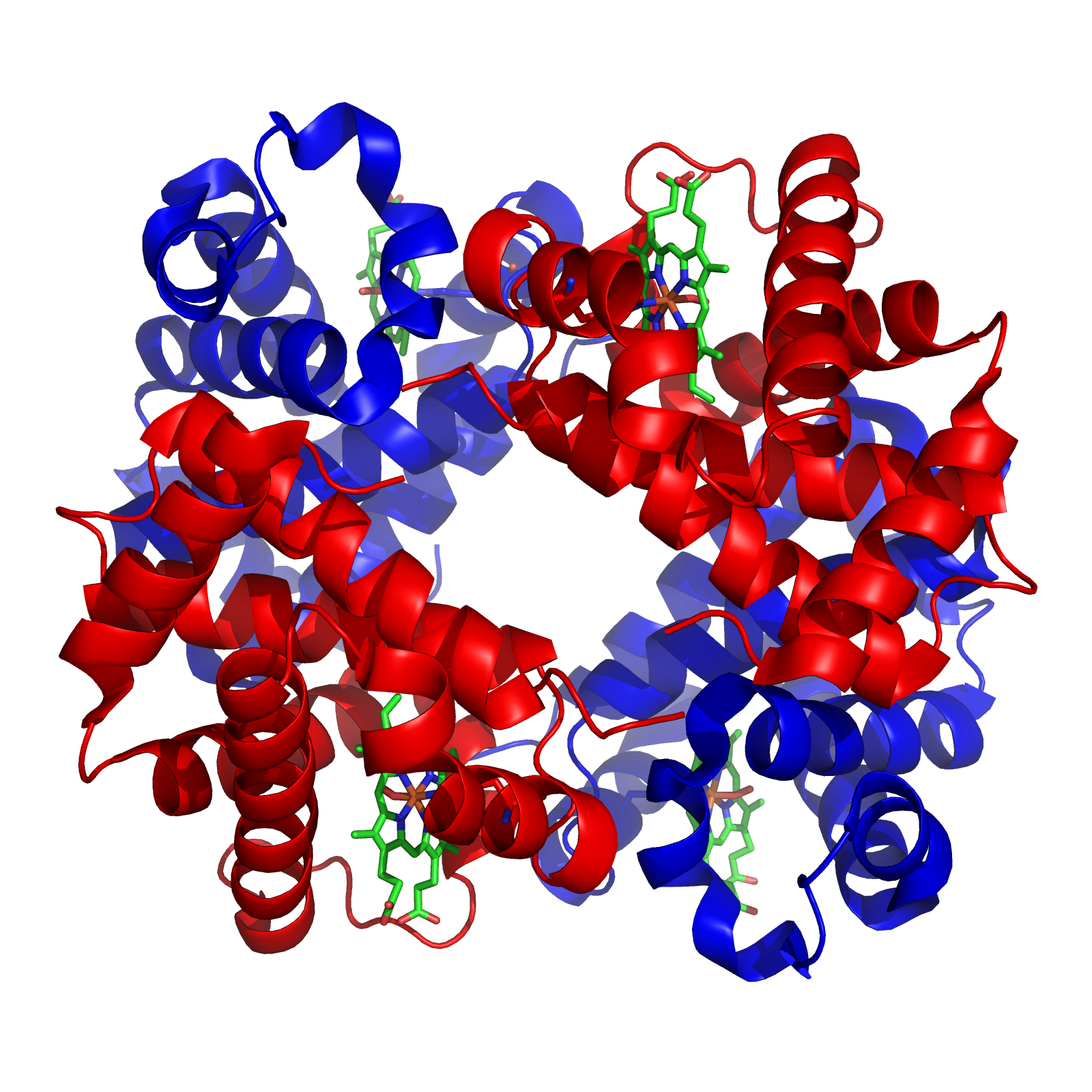 By Richard Wheeler (Zephyris) 2007. Created with p...
By Richard Wheeler (Zephyris) 2007. Created with p... Synthesis of different hemoglobin chains in the pr...
Synthesis of different hemoglobin chains in the pr...