Inborn Diseases
There are many disease considered to be inborn errors of metabolism due to a lack of a particular enzyme. This missing biological catalyst can have many effects on a person. Some of these diseases are Galactosemia, Phenylketonuria (PKU), Lactose Intolerance, and Maple Syrup Urine disease (MSUD).
GALACTOSEMIA
Classic Galactosemia is a rare genetic metabolic disorder. The child with classic galactosemia inherits a gene for galactosemia from both parents, who are carriers. Normally when a person consumes a product that contains lactose (eg dairy products such as milk, cheese, butter) the body breaks the lactose down into galactose and glucose. Glucose is the sugar used by the body for energy. Galactosemia means too much galactose is in the blood caused by the individual "missing" the enzyme (known as GALTA) to convert galactose into glucose. This accumulation of galactose is a poison to the body and can cause serious complications such as an enlarged liver, kidney failure, cataract, and brain damage.
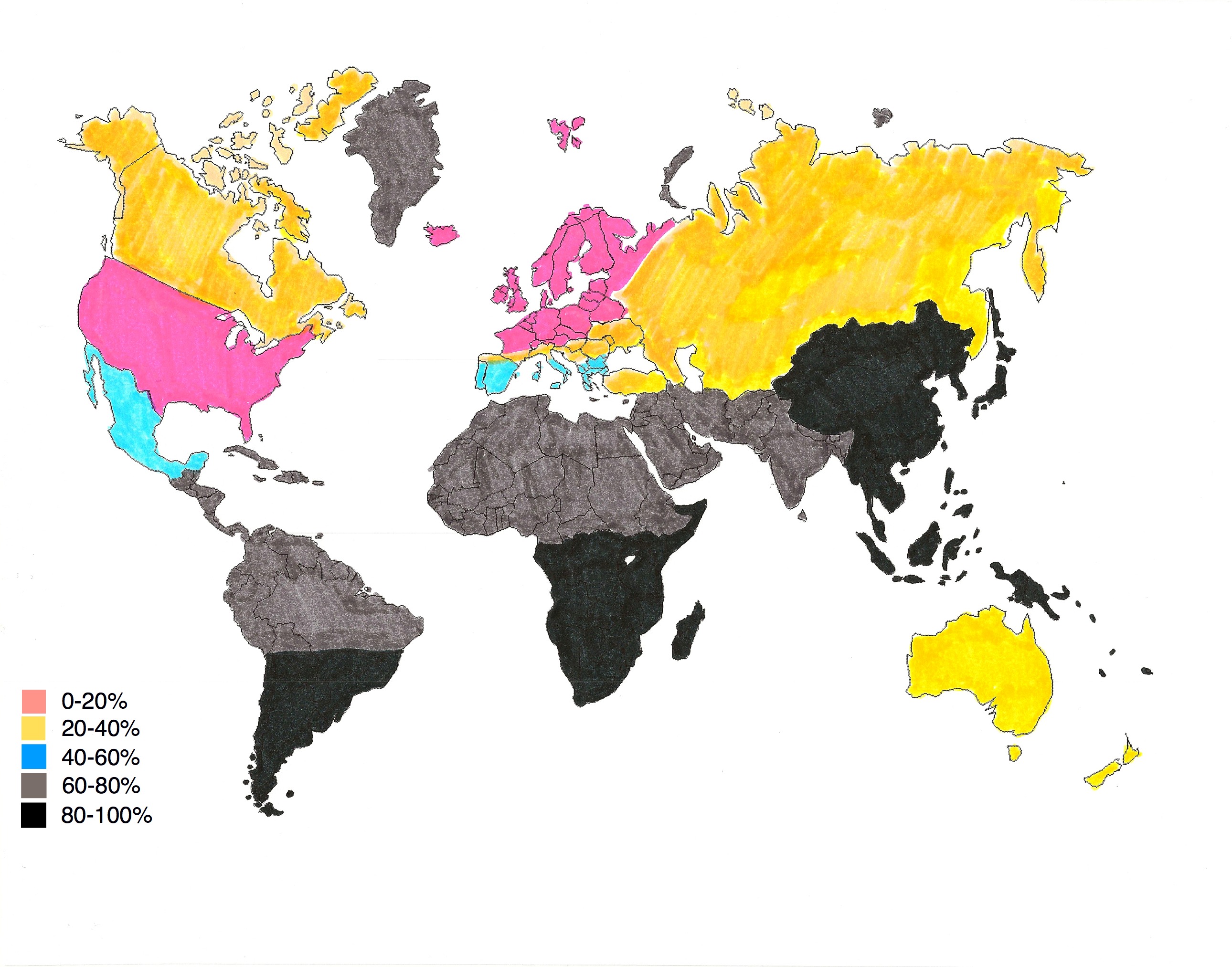 English: Modern-day lactose intolerance by region
English: Modern-day lactose intolerance by region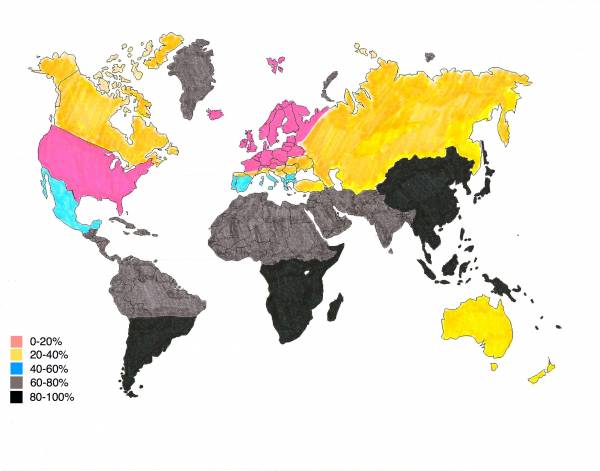 English: Modern-day lactose intolerance in humans
English: Modern-day lactose intolerance in humans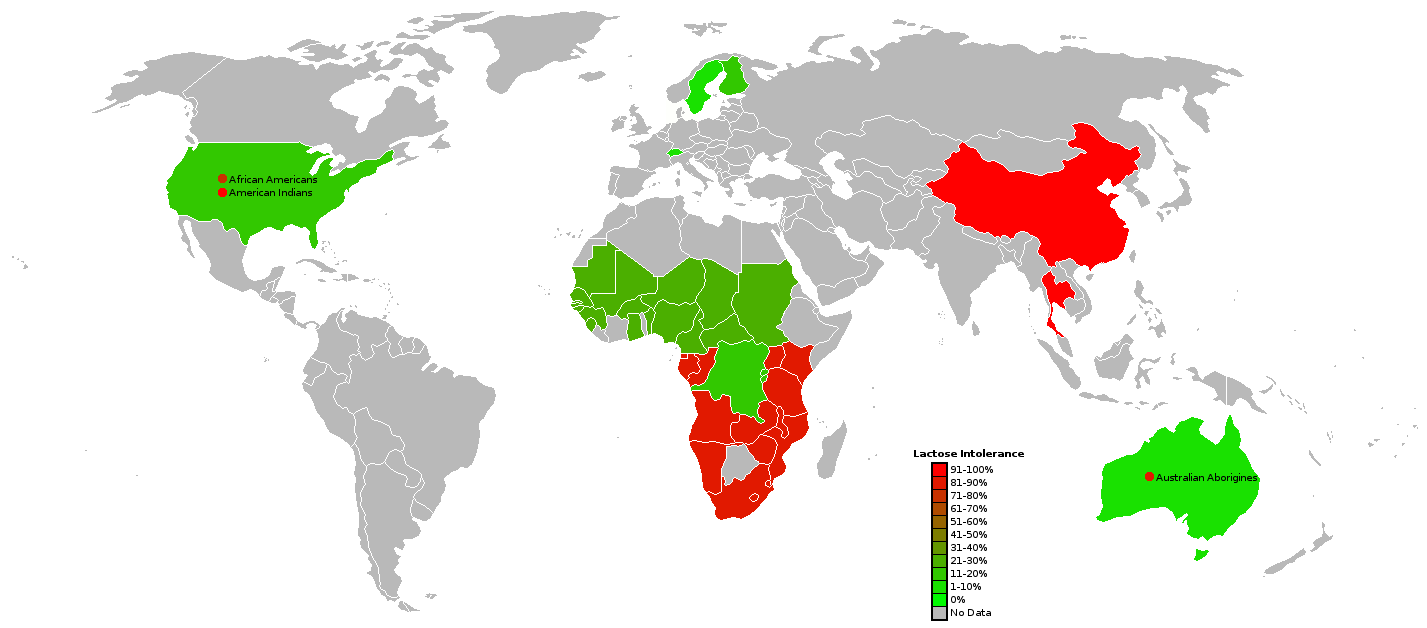 Lactose Intolerance by region of the world (Africa...
Lactose Intolerance by region of the world (Africa...If untreated, as high as 75% of infants will die.
Diagnoisis is made usually within the first week of life by blood test from a heel prick as part of a standrad newborn screening. Treatment requires the strict exclusion of lactose/galactose from the diet. Although galactosemic children are started on diet restriction at birth, there continues to be a high incidence of long term complications involving speech and language, fine and gross motor skill delays and specific learning disabilities. Ovarian failure may occur in girls. Prenatal diagnosis by amniocentesis is also available.
Treatment is based on elimination of galactose from the diet. This may be done in the early neonatal period by stopping breast feeding and by the administratin of diets which contain no lactose or galactose, (Nutramigen, Pregestimil). This diet should be compulsively followed, and continued for years,